
Es ist der 26. April 1986, 0:30 osteuropäischer Zeit. An der nördlichen Grenze der späteren Ukraine – derzeit noch Teil der Sowjetunion – hat Alexander Fjodorowitsch Akimov Bauchschmerzen. Akimov ist Schichtleiter im Reaktorblock 4 des Kernkraftwerks Tschernobyl und betrachtet voller Unbehagen die Anzeigen im Kontrollraum „seines“ Reaktors. Eigentlich sollte er längst damit beschäftigt sein, den Reaktor wieder auf Vordermann zu bringen – aber Genosse Djatlov besteht darauf, den geplanten Test innerhalb der nächsten Stunde durchzuführen. Und wenn der Chef sagt, es werde getestet, dann wird getestet. Denn Akimov ist nicht darauf aus, sich einen neuen Job zu suchen.
Es soll getestet werden, ob der Reststrom, den die Kraftwerksturbinen bei einem Stromausfall liefern, reicht, um die Zeit bis zum Anlaufen der Notstromgeneratoren zu überbrücken. Eigentlich hätte man das schon 1983 tun sollen, bevor man den Kasten für den Regelbetrieb freigab – und nun, nach 3 Jahren Bummelei, hatte man es plötzlich nur allzu eilig damit.
Der „Kasten“ – der Reaktor in Block 4 – besteht aus einem Zylinder aus Graphit – reinem Kohlenstoff – 8 Meter hoch, 12 Meter im Durchmesser, durchzogen von etwa 1700 Kanälen für Brennstäbe, Steuerstäbe und hindurchfliessendes Kühlwasser. Die Anlage ist dazu gedacht, mit einer Nennleistung von 3200 MW Strom zu erzeugen, indem sie bei einer Betriebstemperatur von etwa 300°C – erzeugt durch Kernspaltung – Wasser erhitzt, um mit dem Dampf Turbinen anzutreiben. Der Graphit sorgt dabei als Moderator für die Aufrechterhaltung der Kernspaltungsreaktion.
Einen Gefahren-Test macht man allerdings nicht unter Volllast. Und da man einen Kernreaktor nicht einfach mal schnell runterfahren kann, hat man schon am vergangenen Morgen mit der langsamen Regelabschaltung begonnen. Dann aber brauchten die Genossen unbedingt mehr Strom im Netz, sodass sie noch einen halben Tag bei halber Last weiterproduziert hatten, ehe man weiter herunterfuhr. Und jetzt ist der ganze Kasten voller Xenon-Gift, das die Kernspaltung und damit die ganze Reaktorfunktion ausbremst – so sehr, dass der Reaktor gerade eben wegen irgendeinem Mist beinahe völlig abgeschmiert wäre.
Inzwischen haben Akimov und die Crew nahezu alle Steuerstäbe, die die Kernspaltung bremsen sollen, aus dem Reaktor entfernt, und der Kasten läuft so gerade eben stabil. Von den Vorschriften ist das jedoch weit entfernt! Und das bereitet Akimov umso mehr Bauchschmerzen, je weiter die Vorbereitungen für den Test voranschreiten.
Um 1:23:04 ist es schliesslich soweit: Der Test beginnt. Das Notkühlsystem ist abgeschaltet, damit es im Testverlauf nicht dazwischenfunkt, und Akimov verdrängt den Knoten in seinem Magen, als er die Schliessung der Turbinenschnellschlussventile anordnet – das Startsignal für den Test.
Dadurch wird der Kühlwasserdurchfluss gestoppt, und es wird binnen Sekunden wärmer im Reaktor. Die Wärme fördert die Reaktorleistung ungemein – nun rasch die Bremsstäbe wieder einfahren…wenn die nur nicht so quälend langsam wären! Indessen steigt die Reaktorleistung geradezu exponentiell weiter, denn der anfahrende Reaktor reinigt sich in Sekundenschnelle selbst, während dem Schichtleiter der Schweiss ausbricht: Das gerät ausser Kontrolle – Sofort Abschalten! Akimov betätigt rasch den Notabschaltungsknopf, und alle Bremsstäbe fahren – immer noch langsam! – gleichzeitig in den Reaktor zurück. Doch die Spitzen der Stäbe bestehen aus Graphit, der die Kernreaktion fördert und nicht bremst, bis das eigentliche Bremsmaterial in den Stabschäften tief in den Reaktor gelangt. Doch bis dahin sind die „Bremsen“ wirkungslos: 40 Sekunden nach Testbeginn wird die Kernreaktion endgültig zum Selbstläufer.
So wird es unweigerlich zu heiss im Reaktorkern, und das Ganze fliegt Akimov und der Crew buchstäblich um die Ohren: Wasserleitungen brechen und Wasser kann mit heissem Graphit und Zirkonium aus den Brennstabmänteln reagieren. Es entsteht Wasserstoff, der mit Luft eine explosive Mischung bildet. Eine Knallgas-Explosion deckt schliesslich den Reaktordeckel und das Dach des Kastens ab. Indes fördert Graphit die Kernreaktion umso besser, je heisser er ist. So steigt die Temperatur im Reaktor bis über 2000°C. Die Brennstäbe und alles andere im Reaktorkern beginnen zu schmelzen. Durch das offene Dach kommt dabei Luft an das glühende Riesenbrikett, das vom Reaktor übrig ist, und Hunderte Tonnen Graphit brennen lichterloh. Eine radioaktive Rauchwolke steigt über 1000m hoch aus den Trümmern auf.
Es wird über zwei Wochen dauern, bis allein der Graphitbrand gelöscht und das Austreten des radioaktiven Rauchs unterbunden ist. Akimov erlebt dies nicht mehr. Er stirbt am 11.5.1986 an akuter Strahlenkrankheit.

Der havarierte Reaktorblock 4 am 27. April 1986 – rechts neben dem zerstörten Reaktor sind weitere Schäden am Dach der Turbinenhalle sichtbar. Helikopterpiloten und Fotograf sind während des Überflugs hochgefährlichen Strahlenmengen ausgesetzt. (Bild: Chernobyl NPP)
Wie man aus Atomen Energie gewinnt
Über Atome
Alle Materie der Welt besteht aus Atomen. Jedes Atom besteht aus einem Kern aus Protonen und Neutronen sowie aus einer Elektronenhülle. Auf der Erde sind 118 chemische Elemente bekannt, deren Atome sich durch ihre charakteristische Protonenzahl unterscheiden. Die Neutronenzahl ist hingegen für ein bestimmtes Element nicht festgelegt: Atome eines Elements mit verschiedener Neutronenzahl nennt man Isotope.
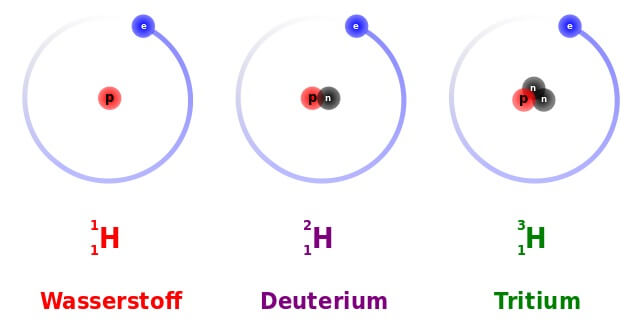
Die drei natürlichen Isotope des Wasserstoffs: Die Zahl links oben in der „Nuklidschreibweise“ steht für die Summe aller Kernteilchen bzw. die Atommasse. Die Zahl links unten steht für die Zahl der Protonen, welche die Zugehörigkeit zu einem Element bestimmt: Atome aller Wasserstoff-Isotope haben ein Proton. Einfacher Wasserstoff (auch „Protium“) und Deuterium sind stabil, Tritium ist radioaktiv. (Bild: Dirk Hünniger (Own work) [GFDL or CC-BY-SA-3.0], via Wikimedia Commons)
Atome können sich zu chemischen Verbindungen – Moleküle, Salze und andere – „zusammentun“, wobei sich ihre Elektronenhülle verändert, die Kerne aber unverändert bleiben (das ist Chemie).
Jedoch können auch Atomkerne verändert werden: Wenn sich dabei die Protonenzahl ändert, entstehen Atomkerne anderer Elemente (das ist Kernphysik bzw. Nuklearphysik).
„Nucleus“ ist übrigens das lateinische Wort für „Kern“. So hat alles, was in der Physik mit dem Begriff „nuklear“ behaftet ist, irgendwie mit Atomkernen zu tun. So hat die Bezeichnung „Nuklear-“ auch Eingang in die Sprache rund um die „Atomenergie“ gefunden.
Energie dank Massendefekt
Die Bildung von Atomkernen aus Protonen und Neutronen funktioniert ähnlich wie die Bildung von Molekülen in der Chemie: Beim Zusammenfügen der Teilchen wird Energie frei. Und je mehr Teilchen im Atomkern zusammenkommen, desto mehr Energie wird frei. Das wird ersichtlich, wenn man Atomkerne wiegt: Ein Heliumkern wiegt nämlich weniger als je zwei einzelne Protonen und Neutronen: Die fehlende Masse wurde als Energie abgegeben! Und da der „zu leichte“ Atomkern zu dem Gedanken verleitet, er sei irgendwie kaputt, nennt man diese Erscheinung „Massendefekt“.
Allerdings gilt das nur für Atomkerne, die höchstens so schwer sind wie ein Eisen-Kern. Um schwerere Kerne als die des Eisens zusammenzubauen, muss man Energie hinzufügen. So sind die Atomkerne der Elemente jenseits des Eisens tatsächlich schwerer als die Summe ihrer Protonen und Neutronen. Findige Physiker kamen so in der ersten Hälfte des 20. Jahrhunderts auf die Idee, schwere Atomkerne auseinanderzubauen, um an diese Energie heranzukommen und sie zu nutzen.
Der Coup mit der Kernspaltung:
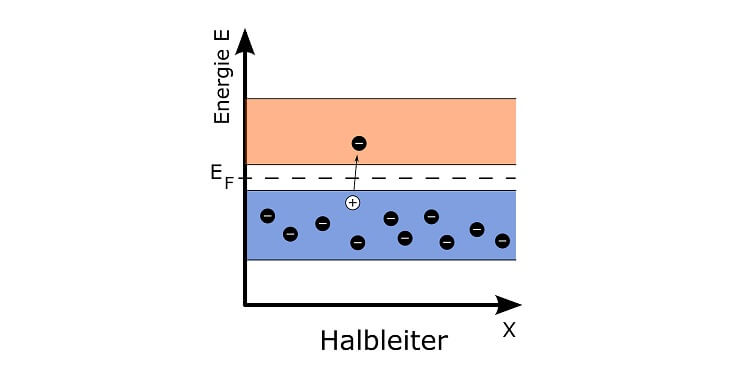
Auf manche Kerne braucht man dazu bloss ein einzelnes Neutron zu schiessen: Sobald das Neutron von solch einem Kern aufgenommen wird, hält dieser nicht mehr zusammen: Er wird gespalten, d.h. er zerfällt in Stücke, darunter meist zwei Kerne leichterer Elemente und ein oder mehrere einzelne Neutronen. Zudem wird ein Teil seiner Kernbindungsenergie frei, teilweise als Bewegungsenergie der Bruchstücke, teilweise in Form von Gamma-Quanten („Licht“) und teilweise in Form grosser Mengen Wärme.
Das Praktische daran ist: Die frei werdenden Neutronen können weitere Atomkerne treffen und spalten, sodass sich die Kernspaltung in einer Kettenreaktion in einem spaltbaren Material unter den richtigen Bedingungen selbst unterhält.
Zu diesen Bedingungen zählt unter anderem die passende Energie bzw. „Geschwindigkeit“ der Neutronen-Geschosse: Zu schnelle Neutronen, wie sie bei einer Kernspaltung freigesetzt werden, prallen nämlich in den meisten Fällen wirkungslos von spaltbaren Kernen ab. Deshalb benötigt man für eine Kettenreaktion neben spaltbarem Material weitere Atome, von welchen schnelle Neutronen abprallen und dabei gebremst – „moderiert“ – werden können. Dafür eignen sich zum Beispiel Wasserstoff-Atome (viele heutige Kernreaktoren enthalten Wasser als „Moderator“), oder Kohlenstoff-Atome, wie Graphit sie enthält.
Nutzung der Kernspaltung: Von Bomben und Steuerstäben
Die Kettenreaktion lässt sich auf zweierlei Weise ausnutzen: Wenn man möglichst viele „langsame“ Neutronen gleichzeitig auf spaltbares Material loslässt, pflanzt sich eine Kettenreaktion in Sekundenschnelle fort und setzt ebenso schnell eine riesige Menge Energie frei, die zu der gewaltigen Explosion einer Atombombe führt.
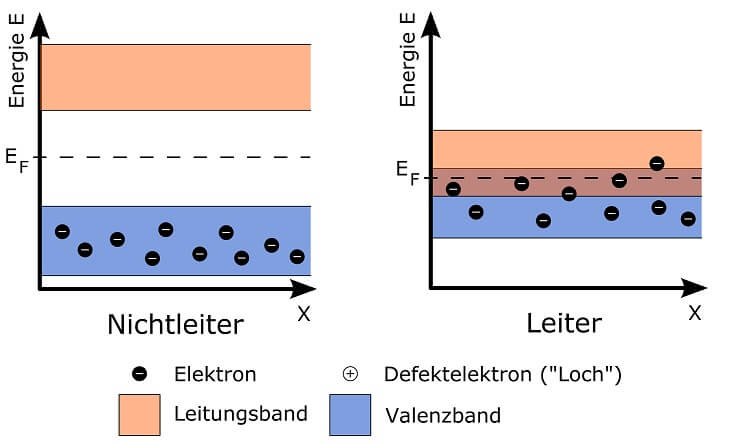
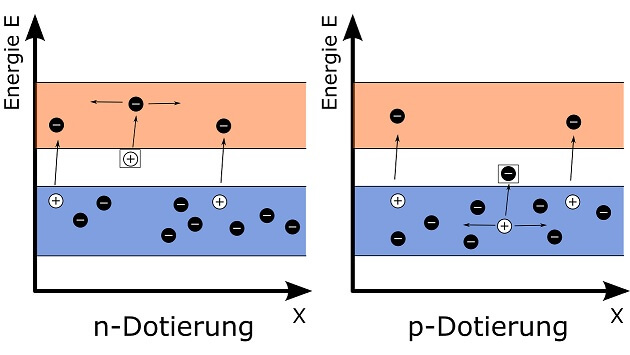
In einer ausgeklügelten Anlage kann man hingegen die Menge der zur Kernspaltung nutzbaren Neutronen sehr genau steuern. Dazu verwendet man bewegliche „Steuerstäbe“ aus einem Material, dessen Atome Neutronen aufnehmen können ohne gespalten zu werden. Je weiter diese Stäbe in einen Block aus spaltbarem Material und Moderator eingebracht werden, desto mehr Neutronen werden „verschluckt“ und können nicht mehr an der Kettenreaktion teilhaben. Mit solch einem Reaktor, in dem kontrolliert Wärme durch Kernspaltung entsteht, kann man in einem Atomkraftwerk Strom erzeugen.
Und was ist mit der Strahlung?
Obwohl herumliegende Trümmer eindeutig eine andere Sprache sprechen, beharrt die Kraftwerksleitung in Tschernobyl bis zum Abend des 26. Aprils darauf, dass der Reaktor bei dem Unglück intakt geblieben sei. In Folge dessen wird die Bevölkerung der 5 km entfernten Siedlung Prypjat erst am 27. April evakuiert.
Erst, nachdem im 1200 km entfernten schwedischen Kernkraftwerk Forsmark am 28. April um 9:00 wegen erhöhter Radioaktivität Alarm ausgelöst wird, welche nach erfolgloser Suche nach eigenen Lecks auf einen Fallout aus (Wind-)Richtung Sowjetunion zurückgeführt wird, dringt die Nachricht von dem Unglück in Tschernobyl in den Westen durch.
Die Nachrichten von radioaktivem Niederschlag über weiten Teilen Europas schüren Verunsicherung und Ängste bei der Bevölkerung. Allein in Nordrhein-Westfalen werden Hunderte Naturwissenschaftler – „jeder, der irgendein chemisches Element buchstabieren konnte“ – rekrutiert, um die Notfall-Hotline der Landesregierung zu besetzen und die Fragen zahlloser verängstigter Bürger zu beantworten.
Ich bin viereinhalb Jahre alt, als mein Vater – einer der Physiker, die an jener Hotline Dienst taten – uns erklärt, dass Mama meine Schwester und mich nicht im Garten spielen lasse, weil es nach einem schlimmen Unfall weit im Osten „Gift“ geregnet habe.
Inzwischen sind die ersten der insgesamt 600.000 bis 800.000 „Liquidatoren“ in Tschernobyl mit Aufräumarbeiten beschäftigt: Jene Männer, die Trümmer des zerstörten Block 4 vom Dach des benachbarten Blocks 3 räumen, dürfen sich der Strahlung wegen nur jeweils 40 Sekunden auf dem Dach aufhalten. Ein Mann für jede Schaufel Abraum. Andere werfen mit Hubschraubern Löschmittel auf den havarierten Reaktor ab, begraben tonnenweise Erde unter Beton, versuchen Staub an den Boden zu binden. Das Gebiet im Radius von über 30 Kilometern um das Kernkraftwerk wird evakuiert, dort lebende Tiere getötet, damit sie die Strahlung nicht aus der Sperrzone hinaustragen…
Aber was ist „radioaktive Strahlung“ eigentlich, und warum ist sie so gefährlich?
Was ist Radioaktivität?
„Radioaktivität“ ist eine Eigenschaft einiger Atomkerne: Nicht alle Kombinationen von Protonen und Neutronen in einem Atomkern halten fest zusammen. Solche instabilen Kerne sind radioaktiv, sie „bröckeln“: Früher oder später löst sich ein „Bröckel“ aus dem Atomkern und fliegt – sofern er nicht von aussen beeinflusst wird – mit Geschwindigkeiten im Bereich von 10000 km/s [2] geradeaus davon.
Solche „Bröckel“ können α- oder β-Teilchen sein (da diese Teilchen geradlinig vom Atomkern wegfliegen, erscheinen sie auf den ersten Blick wie „Strahlen“ und werden oft auch so genannt). α-Teilchen sind nackte Helium-Atomkerne, bestehend aus je zwei Protonen und Neutronen, während β-Teilchen Elektronen (oder Anti-Elektronen, „Positronen“) sind, die entstehen, wenn ein Neutron im Kern zu einem Proton zerfällt und dabei ein Elektron abgibt (bzw. ein Proton ein Positron abgibt und als Neutron verbleibt). Die einzigen „echten“ Strahlen sind γ-Strahlen: Dabei handelt es sich um sehr energiereiche „Licht“-Wellen, die von besonders energiereichen Atomkernen abgegeben werden können.
In der Natur gibt es viele derart instabile Kerne, die radioaktiv sind. Viele Elemente bestehen aus einem Gemisch aus stabilen und radioaktiven Isotopen, sodass Stoffe, die sich aus solchen Elementen zusammensetzen, automatisch diese verschiedenen Isotope enthalten. So findet man zum Beispiel in Bananen, die von Natur aus viel Kalium enthalten, auch Atome des radioaktiven Isotops 40K, und jeder Mensch, der naturgemäss aus Kohlenstoffverbindungen besteht, enthält Atome des radioaktiven Isotops 14C. Dass unsere Umgebung einschliesslich uns selbst „strahlt“, ist also erst einmal normal.
Einmal abgestrahlte α-Teilchen kommen jedoch nicht weit: Auf ihrem Weg stossen sie immer wieder gegen andere Teilchen und verlieren an Energie und damit an Geschwindigkeit. Schon nach ein bis zwei Millimetern Flugstrecke in Luft ergattern sie sich irgendwo zwei Elektronen und werden zu normalen Helium-Atomen. β-Teilchen (Elektronen) finden spätestens nach 10 Metern in Luft ein Atom als neue „Heimat“. γ-Strahlen verhalten sich hingegen wie fast alles durchdringendes Licht und breiten sich geradezu unendlich weit aus, wenn ihnen keine besonders dichte Materie, wie ein dicker Blei-Klotz, im Wege steht.
Das „Ende“ von α- und β-Teilchen deutet es schon an: Wenn solch ein reisender „Bröckel“ mit Volldampf auf ein Atom trifft, kann dieses „kaputtgehen“: Der Einschlag kann Elektronen aus der Hülle schleudern. Die „Strahlen“ wirken ionisierend (auch γ-Strahlen haben diese verheerende Wirkung: Sie sind energiereicher als Licht (Lichtphänomene) und regen Elektronen so stark an, dass diese „ihr“ Atom verlassen können!).
Der Begriff „Ionisierende Strahlung“ beschreibt diese herumfliegenden Atomtrümmer also besser als der Pleonasmus „radioaktive Strahlung“ („radioaktiv“ bedeutet nichts anderes als „strahlend“).
Wie misst man „radioaktive“ bzw. ionisierende Strahlung?
Die ersten Wissenschaftler, die sich mit Radioaktivität beschäftigten, ahnten noch nichts von ihrer Gefährlichkeit. So interessierten sie sich vornehmlich für die Menge der Strahlung, die von einem radioaktiven Stoff ausging. Die Curies „erfanden“ deshalb die später nach ihnen benannte erste Masseinheit für die Aktivität – den Vergleich mit der Aktivität von einem Gramm Radium.
In einem Gramm Radium zerfallen in jeder Sekunden 37 Milliarden Atome und geben „Strahlen“ ab. Das entspricht einer Aktivität von einem Curie (Ci). Dieser enormen Strahlungsmenge sollte man jedoch tunlichst fern bleiben. So wird heute eine wesentlich „handlichere“ Einheit für die Aktivität verwendet:
Ein Gramm Radium hat also eine Aktivität von 37 Milliarden Becquerel! Ein Gramm Natur-Uran hätte hingegen eine Aktivität von 25.290 Becquerel, ein Gramm natürliches Kalium 31,2 Becquerel. [1]
Wer sich mit der Gefährlichkeit von ionisierender Strahlung beschäftigt, wird sich allerdings mehr dafür interessieren, wie viele Strahlen einen Menschen (oder anderen Organismus) tatsächlich treffen und in ihm Schaden anrichten. Und Schaden wird angerichtet, wenn die Atome des Körpers die (Bewegungs-)Energie einfallender Strahlung aufnehmen. Deshalb wird häufig eine Energiedosis für ionisierende Strahlung angegeben:
Wir alle sind tagtäglich natürlicher ionisierender Strahlung aus dem Weltraum ausgesetzt. Jedes Kilogramm unserer Körper nimmt daher täglich 3*10-5 (drei Hunderttausendstel) Gray aus der Weltraumstrahlung auf [2]. Die 1000 Liquidatoren, die am ersten Tag nach dem Tschernobyl-Unglück in unmittelbarer Nähe von Reaktorblock 4 eingesetzt wurden, bekamen dort eine Energiedosis von etwa 2 bis 20 Gray ab [1].
Strahlung ist aber nicht immer gleich Strahlung. Wenn ein α-Teilchen mit hoher Geschwindigkeit auf ein Atom trifft, kommt das einem nuklearen Crash mit einem Lastwagen gleich, während sich die Begegnung mit einem ähnlich schnellen, aber rund 1000 mal leichteren β-Elektron im Vergleich dazu wie der Zusammenstoss mit einem Radfahrer ausnimmt. Zur Bestimmung der Gefährlichkeit der Strahlenarten muss ihre Energiedosis daher mit einem „Gefährlichkeitsfaktor“ multipliziert werden. Wenn dieser Faktor für β-Teilchen und γ-Strahlen 1 ist, beträgt er für α-Teilchen 20.
Pro Jahr ist ein Mensch durchschnittlich zwei Tausendstel Sievert (2 mSv) aus natürlicher Strahlung ausgesetzt, in Gegenden mit besonderen Vorkommen radioaktiver Elemente im Boden sogar deutlich mehr. Im Fall einer kurzzeitigen(!) Begegnung mit starker Strahlung macht sich eine Dosis bis etwa 200 mSv durch keinerlei Symptome bemerkbar. Erst darüber treten Symptome der Strahlenkrankheit auf. Wenn Menschen in kurzer Zeit einer Strahlendosis von 4,5 Sv oder mehr ausgesetzt sind, stirbt jeder zweite innerhalb von vier Wochen. Eine Dosis von 6 Sv oder mehr in kurzer Zeit gilt als absolut tödlich – die gleiche Dosis innerhalb von 50 Jahren bleibt hingegen ohne messbare Folgen [2].
Was bewirkt ionisierende Strahlung?
Ionisierende Strahlung kann mit einem Geigerzähler (eigentlich: Geiger-Müller-Zählrohr) registriert werden: In dem Zählrohr befindet sich ein dünnes Gas aus Atomen, die von einfallender ionisierender Strahlung in Ionen und Elektronen gespalten werden und in einem elektrischen Feld zu zwei Polen hingezogen werden. Die wandernden geladenen Teilchen schliessen so einen Stromkreis, was sich im angeschlossenen Lautsprecher als „Knack“ bemerkbar macht. Je mehr „Knacks“ es gibt, desto mehr ionisierende Strahlen sind in das Gas im Zählrohr eingeschlagen.
Wenn die ionisierende Wirkung ein Atom in einem Molekül trifft (zum Beispiel in einem Biomolekül wie DNA), kann das Molekül als solches Schaden nehmen. Da Radioaktivität eine ganz natürliche Sache ist, haben Zellen – auch menschliche – verschiedene Mechanismen entwickelt, um kaputte Biomoleküle, insbesondere DNA, bei Bedarf zu reparieren. Erst wenn die Zellen mehr ionisierende „Treffer“ einstecken müssen, als sie reparieren können, entstehen spürbare Zell- und Gewebeschäden.
Bei sehr grossen Strahlenmengen äussert sich das als „Strahlenkrankheit“. Weniger grosse oder über längere Zeit ertragene Strahlenmengen spürt man hingegen nicht sofort – was sie so tückisch macht. Dauerhaft beschädigte DNA kann jedoch – auch lange nach der Begegnung mit der Strahlung – zu Erkrankungen wie Krebs und Leukämie führen.
Was hat Radioaktivität bzw. ionisierende Strahlung mit Atomkraftwerken zu tun?
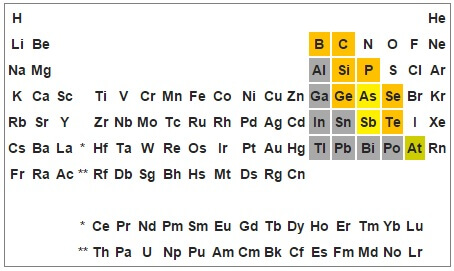
Die wohl wichtigste Atomsorte, die für die Kernspaltung geeignet ist und in grösseren Mengen in der Natur vorkommt, ist das Uran-Isotop 235U. Deshalb findet 235U sowohl in ersten Atombomben als auch in Reaktoren Verwendung (eigentlich ist das Isotop 238U noch sehr viel häufiger, aber nicht spaltbar, sodass bei der Herstellung von Kernbrennstoff ein Teil des 238U aufwändig vom Rest getrennt werden muss, um für die Kettenreaktion ausreichend „anzureichern“).
Unglücklicherweise sind sowohl 235U als auch 238U von Natur aus radioaktiv. Beide Isotope sind α-Strahler, d.h. sie zerfallen zu Heliumkernen und Isotopen des Elements Thorium – die wiederum radioaktiv sind. Die Halbwertszeit – also jene Zeitspanne, in welcher die Hälfte einer Portion einer Atomsorte zerfällt, beträgt für 235U 703,8 Millionen Jahre, für 238U 4,47 Milliarden Jahre (das entspricht etwa dem Alter der Erde!). Eine Portion Uran enthält also immer – grossteils radioaktive – Atome einer ganzen Reihe verschiedener Elemente, die im Zuge der Abfolge verschiedener Zerfälle entstehen.
Allein deshalb erfordert der Umgang mit Uran schon besondere Sicherheitsvorkehrungen. Der eigentliche Haken an der Sache kommt aber noch:
In den wenigen Wochen, die das Uran in einem Kernreaktor zubringt, entsteht eine Vielzahl von Spaltprodukten und sehr schweren Atomkernen, die oftmals ihrerseits radioaktiv sind, durch „Verschlucken“ der herumfliegenden Neutronen.
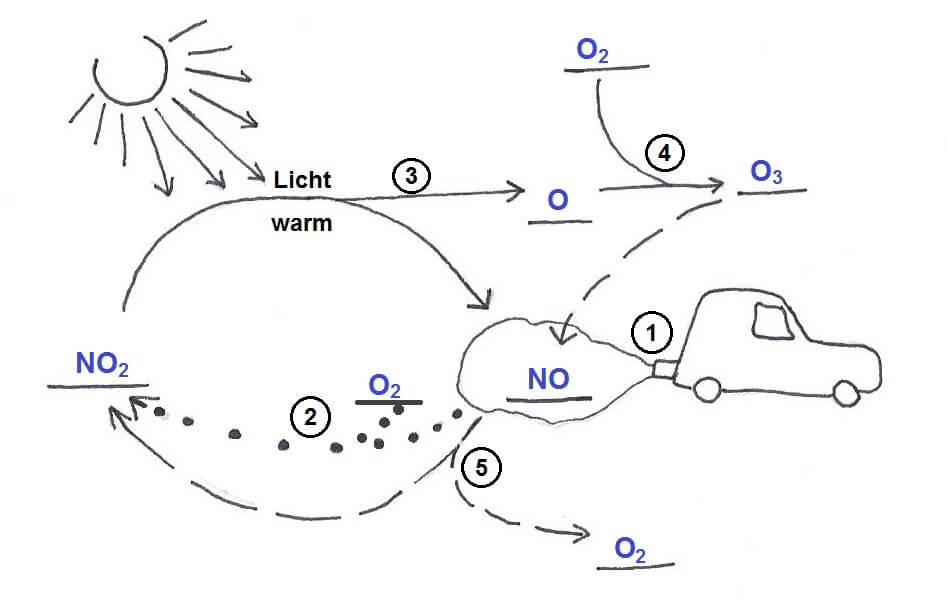
Zu den Spaltprodukten zählt zum Beispiel das Xenon-Isotop 135Xe, das mit Vorliebe Neutronen schluckt und die Kettenreaktion ausbremst. Deshalb wird dieses Isotop als „Reaktor-Gift“ (das ist der „Xenon-Müll“ in der Einleitung) bezeichnet. Wenn ein Reaktor unter Volllast läuft, reagiert das 135Xe jedoch ebenso schnell weiter, wie es entsteht, sodass es sich nicht ansammelt. Während der Reaktor in Tschernobyl einen halben Tag lang nur mit halber Kraft lief, ist hingegen mehr 135Xe entstanden als abgebaut werden konnte, was die Kettenreaktion regelrecht ausgebremst und zu dem dramatischen Leistungseinbruch vor Beginn des fatalen Tests geführt hat.
Das Berüchtigste unter den entstehenden schweren Elementen, auch „Transurane“ genannt, ist das Plutonium, dessen Isotop 239Pu ebenfalls spaltbar ist und sowohl als Reaktor-Brennstoff als auch für Atombomben taugt (deshalb werden Uran-Brennstäbe gemäss den Regeln der Internationalen Atomenergie-Behörde so lange im Reaktor belassen, bis das entstehende 239Pu zu nicht spaltbarem 240Pu weiterreagiert ist [2]).
Insgesamt strahlt die bunte Mischung von Atomkernen in „verbrauchten“ Brennstäben rund 10 Millionen mal stärker als „frisches“ Uran! Und dabei haben viele dieser Kerne solch lange Halbwertszeiten, dass die Brennstäbe selbst 10 Jahre nachdem sie ausrangiert wurden, noch rund eine Million mal stärker als „frisches“ Uran strahlen [2]. Daraus ergibt sich die ungeheure Problematik bei der Lagerung dieses „Atommülls“: „Verbrauchtes“ Brennmaterial sollte möglichst lange möglichst weit weg bzw. abgeschottet von allem lagern können, bis seine radioaktiven Bestandteile zerfallen sind. Und da „möglichst lange“ viele Jahrtausende meint, gibt es noch keine Technologie, welche eine gefahrlose Lagerung auf der Erde über so lange Zeit wirklich sicherstellt.
Leser fragen zu Kernkraft und Radioaktivität:
Cornel van Bebber fragt auf Google+:
Was ist der Unterschied zwischen dem radioaktiven Zerfall im Kernkraftwerk in Tschernobyl und der Atombombe?
Im Grunde genommen gibt es keinen – denn der Brennstoff im Reaktor und Atombomben bestehen aus den gleichen Stoffen (allerdings muss das spaltbare Material für den Bau einer funktionierenden Bombe um einiges stärker angereichert werden als für den Kraftwerksbetrieb). Die Reaktionen, welche in einem Kraftwerk innerhalb von Wochen und Monaten ablaufen, finden bei der Explosion einer Atombombe in Sekunden statt – die Spalt- und Nebenprodukte sind aber weitestgehend die gleichen.
Besonders berüchtigt sind vor allem Cäsium-137 (137Cs), ein β-Strahler mit einer Halbwertszeit von 30 Jahren, und Iod-131 (131I), ein β-Strahler mit einer Halbwertszeit von 8 Tagen. Beide sind mögliche Bruchstücke, die bei der Spaltung von 235U entstehen, und somit sowohl aus einer Bombe als auch aus einem havarierten Reaktor freigesetzt werden können.
Cäsium-137 ist besonders gefährlich, weil es chemisch den anderen Alkalimetallen, vor allem dem Kalium, ähnelt und leicht an deren Stelle einen Weg in Organismen findet, und weil es zu energiereichen, „angeregten“ Barium-137-Kernen zerfällt, die wiederum γ-Strahlen abgeben, um zu nicht-strahlendem Barium-137 ( 137Ba ) zu werden. In der Medizin macht man sich diese γ-Strahlen bei der Strahlentherapie von Tumoren zunutze, aber in „freier Natur“ können sie auch auf gesundes Gewebe eine verheerende Wirkung haben.
Iod-131 zerfällt zwar recht schnell, aber der menschliche Körper lagert Iod-Atome rasch in der Schilddrüse ein, um sie zu Hormonen verarbeiten zu können. Dabei unterscheidet der Organismus nicht zwischen verschiedenen Isotopen, da diese sich chemisch gleichen. So kann in der Schilddrüse gesammeltes Iod-131 in kurzer Zeit merkliche Schäden anrichten und beispielsweise Schilddrüsenkrebs auslösen. Deshalb sind in der Schweiz im Einzugsgebiet von Kernkraftwerden Iod-Tabletten mit nicht-strahlendem Iod ausgegeben worden, die die Bevölkerung im Falle einer Freisetzung von Iod-131 schnell einnehmen soll. Damit soll erreicht werden, dass die betroffenen Körper das Tabletten-Iod zuerst einlagern und für Iod-131 möglichst keinen Platz mehr lassen.
Der grosse Unterschied zwischen Bombe und Kernkraftwerk besteht letztlich darin, dass das radioaktive Material durch die Explosion einer Atombombe weit verteilt wird und schnell ein grosses Gebiet „verstrahlt“, während es im Kernkraftwerk samt seiner abgestrahlten Teilchen und Strahlen im Reaktor bleibt und niemandem direkt schadet – normalerweise jedenfalls.
Nach dem Unglück in Tschernobyl durften meine Schwester und ich unseren Physiker-Vater in den Garten begleiten, um Bodenproben aus dem Sandkasten, Mamas Beeten und vom Grund des Gartenteichs zu nehmen. Mit den Proben sind wir dann nach Düsseldorf in die Uni gefahren, wo es eine Zähl-Apparatur gab, mit welcher Papa die Strahlung aus dem „Gift“ in unserem Garten messen und – so hofften wir zumindest – das Draussen-Spiel-Verbot allenfalls wieder aufheben konnte. Unsere und andere Messungen von Papas Kollegen ergaben allerdings, dass von den Böden in der Umgebung doch um einiges mehr Strahlung ausging als normal gewesen wäre.
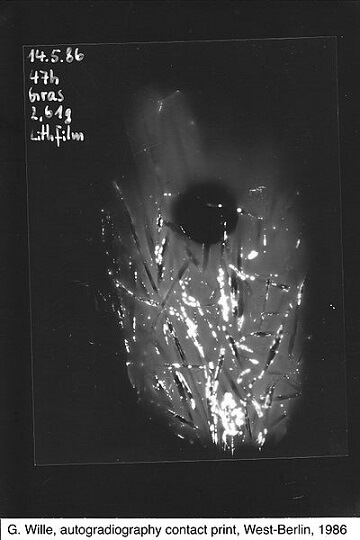
Schüler in West-Berlin legten am 15. Mai 1986 ein Büschel Gras von ihrem Schulhof für knapp 2 Tage auf einen Planfilm. Ionisierende Strahlung schwärzt, vergleichbar mit Licht, Filmmaterial. Auf dem hier gezeigten Negativ erscheinen stark strahlende Bereiche weiss. Der runde, schwarze Fleck rührt von einer Münze her, welche die gestreute Strahlung abschirmt. Wenn die weissen Flecken hier tatsächlich, wie von den Autoren angegeben, von Iod-131 herrühren, ist dem Datum nach davon auszugehen, dass hier allenfalls ein Viertel des ursprünglich in diesem Fallout freigesetzten Iod-131 „detektiert“ wurde (bei einer Halbwertszeit von 8 Tagen hat sich die Menge des Isotops seit der Freisetzung schon zweimal halbiert). (Bild: ViolaceinB (Own work) [CC BY-SA 4.0], via Wikimedia Commons)
Cedric97 von itscedric.de fragt:
Block 4 ist ja in die Luft geflogen, aber die anderen drei Reaktoren liefen ja noch Jahre weiter. Meine Frage: Warum wurden die anderen Reaktoren weiter betrieben?
Laut der Wiener Umweltanwaltschaft, die auf Wikipedia zitiert wird, hat das Kernkraftwerk Tschernobyl – für die Sowjetunion eine „Vorzeige-Anlage“ – seinerzeit ein Sechstel des Atomstroms auf dem Gebiet der Ukraine geliefert, was 4 bis 10% des Gesamtstroms entspricht. Darauf konnte oder wollte der Staat seinerzeit nicht von jetzt auf gleich verzichten. Tatsächlich war die Fertigstellung der im Bau befindlichen Blöcke 5 und 6 nach Absinken der Radioaktivität noch geplant. Erst nach dem Zusammenbruch der Sowjetunion konnte die Regierung der seither unabhängigen Ukraine gegen Ausgleichszahlungen seitens der EU dazu bewegt werden, die verbliebenen Blöcke 1 bis 3 des Kraftwerks endgültig abzuschalten („Memorandum of Understanding“ zwischen den G7-Staaten und der Ukraine vom 20.12.1995).
Gibt es bereits Vergleiche mit Tschernobyl zu den beschädigten Kernkraftanlagen in Belgien?
In meinen Augen wäre ein solcher Vergleich gar nicht sinnvoll: Die Kernkraftwerke Doel und Tihange in Belgien arbeiten mit Druckwasser-Reaktoren. Darin wird Wasser als Moderator und Wärmeüberträger verwendet, welches durch Überdruck im Reaktor-Druckbehälter am Verdampfen gehindert wird. Diese Druckbehälter einer bestimmten Baureihe weisen in Belgien nun eine grosse Anzahl Haarrisse auf. Würde ein solcher Behälter Leck schlagen, sodass es zu einem Druckabfall kommt, könnte das Wasser darin verdampfen und seine wärmeabführende Wirkung verlieren. Dann bestünde die Gefahr einer Kernschmelze.
Der Tschernobyl-Reaktor vom Typ RBMK-1000 war hingegen ein Siedewasser-Druckröhrenreaktor, der statt einem Druckbehälter viele unter Druck stehende Röhren mit Brennstoff sowie Graphit als Moderator enthielt. Auch weitere Sicherheitshüllen („Containment“), wie sie die belgischen (und andere westliche) Reaktoren umgeben, haben die RBMK-Reaktoren nicht. Die Freisetzung des hoch radioaktiven Materials aus dem Reaktor-Kerns nach der einmal eingetretenen Kernschmelze ist demnach nicht zuletzt auf die baulichen Schwächen dieses Reaktor-Typs zurückzuführen.

Geschmolzenes radioaktives Material aus dem havarierten Reaktor ist im Keller von Block 4 in Tschernobyl aus einem Ventil zur Dampf-Ableitung ausgetreten und erstarrt. Hier gab es keine Sicherheitsbehälter, die die lavaartige Schmelze hätten zurückhalten können. (Bild: The Kurchatov Institute (Russia) and the ISTC-Shelter (Ukraine); Quelle: International Nuclear Safety Program)
Als Nicht-Kernkraft-Ingenieurin kann ich also nur hoffen, dass die Verantwortlichen in Belgien (wie auch in der Schweiz – das Kernkraftwerk Mühleberg verwendet einen Reaktor-Druckbehälter des gleichen Herstellers wie die Kraftwerke in Belgien) wissen, was sie tun, und sich anders als die Ingenieure in Tschernobyl an ihre Vorschriften halten, bis die betreffenden Reaktoren endgültig vom Netz genommen werden.
Renate Thormann schreibt auf Facebook:
Ich halte es für wichtig, dass man sich immer und immer wieder der Langzeitfolgen bewusst bleibt. Da wird nicht genug hingesehen und es ist auch mangels Erfahrung gar nicht bekannt, was alles geschieht, wenn man lange auf kontaminiertem Gebiet lebt. Im Staate Washington ist grade wieder eine Riesenkatastrophe mit nuklearem Material geschehen. Das Leck dort bestand schon seit 2011 .. jetzt leckt es mehr als massiv, nämlich katastrophal. Was hört man davon? Nix? Eben … darum finde ich es am wichtigsten so viel wie möglich zu informieren. Langzeitfolgen im Auge zu behalten und Erfahrungen mit Radioaktivität auszutauschen. Es gibt eine „Sarkophag“ Seite, die immer über Tschernobyl berichtet ..
Innerhalb von 206 Tagen nach der Havarie von Tschernobyl umschliessen rund 90.000 Liquidatoren den gesamten Reaktorblock 4 mit einem zwanzig „Stockwerke“ hohen Kasten aus Stahlbeton, in dem die verbliebenen Überreste des geschmolzenen Kerns seither ruhen wie in einem Sarkophag.
Dieses schon gewaltige Bauwerk war auf eine Lebensdauer von etwa 30 Jahren ausgelegt, doch der Zahn der Zeit zeigte seine Spuren schon weitaus früher – es gibt verschiedene Berichte von Undichtigkeiten oder gar Teil-Einstürzen. So ist im Augenblick ein zweiter Sarkophag im Bau, welcher noch über den ersten geschoben werden und 100 Jahre halten soll. Doch was kommt dann?

Der neue Sarkophag im März 2016: Nach seiner Fertigstellung soll er über den alten Sarkophag (links im Hintergrund) geschoben werden. Das macht ihn zum bis Dato grössten beweglichen Gebäude der Welt. (Bild: Tim Porter (Eigenes Werk) [CC BY-SA 4.0], via Wikimedia Commons)
Und die Reaktor-Ruine von Tschernobyl ist nicht die einzige unnatürliche Quelle ionisierender Strahlung. Moderne, nach allen Sicherheitsvorschriften betriebene Kernkraftwerke zählen meines Wissens nicht dazu. Stattdessen sind es Altlasten, die zunehmend Sorgen bereiten. Da ist die von Renate erwähnte Hanford Site in Washington State im Nordwesten der USA, ein ehemaliges Versuchsreaktoren-Gelände, auf welchem grosse Mengen radioaktiver Abfälle in Tanks lagern – Tanks, die altern und zunehmend Lecks aufweisen. Da ist der Reaktorunfall in Fukushima in Japan, der letztlich von Naturgewalten ausgelöst wurde. Die austretende Strahlung ist in der Natur dennoch nicht vorgesehen. Da sind die über Hiroshima und Nagasaki eingesetzten Atombomben und zahllose weitere Atomwaffentests
Nichts desto trotz ist das Atomzeitalter, wenngleich es sich nach der Meinung vieler dem Ende neigen sollte, noch jung – es zählt weniger als 100 Jahre. So sind unsere Kenntnisse der Langzeitfolgen durch das Tschernobyl-Unglück und anderer unnatürlicher Strahlenquellen bestenfalls lückenhaft, in mancher Hinsicht noch gar nicht abzusehen, und sie werden nach wie vor kontrovers diskutiert.
So schliesse ich mich Renate von Herzen an: Erinnern wir uns an jene schicksalhafte Nacht auf den 26. April 1986, und an alle anderen schicksalhaften Augenblicke des Atomzeitalters und behalten wir ihre Folgen im Auge, um daraus für die Zukunft zu lernen.
Dieser Post enthält (nicht nur) meine Erinnerung und mag hoffentlich helfen zu verstehen, worum es bei Atomen, Kernkraft und Radioaktivität eigentlich geht.
Erinnert ihr euch noch an das Tschernobyl-Unglück? Wie habt ihr jene Tage vor 30 Jahren erlebt? Oder seid ihr erst „nachher“ zur Welt gekommen? Welche Bedeutung haben die Ereignisse für euch?
Literatur:
[1] Es gibt einen ausführlichen Artikel zum Unglück von Tschernobyl auf Wikipedia: https://de.wikipedia.org/wiki/Nuklearkatastrophe_von_Tschernobyl. Die Einleitung des Posts ist eine freie Nacherzählung anhand der dortigen Darstellung des Unfallhergangs.
[2] Rudolf Kippenhahn (1998): Atom.Forschung zwischen Faszination und Schrecken. Erweiterte Taschenbuchausgabe im Piper-Verlag GmbH, München. (Das Buch ist vergriffen, aber auf dem Gebrauchtmarkt und in Bibliotheken zu finden: Ein umfassendes, auch für den Laien verständliches Werk, das durch die Geschichte der Atome und ihrer Erforschung führt und schliesslich umfassende Informationen zu Kernenergie und Radioaktivität bereithält.
[3] Weitere Einzelheiten und Bilder rund um Tschernobyl und das Unglück in englischer Sprache gibt es auch auf http://chernobylgallery.com/




















{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}